Bumping the NGS read classification rate to 97%
Last year, we published our paper on two-step metagenomic classification. The next stage of this work is now starting to bear fruit. In experiments, we have seen the read classification rate of randomly selected SRA samples go up from 74% to 97%.
First, some background. Kraken 2 is one of the most popular metagenomic classification and profiling (with Bracken) tools. Our contribution is to reimplement this algorithm as a distributed system on Apache Spark, making it much more scalable, and also a new process that we call two-step classification. We call our tool Slacken.
Kraken 2 is distinguished from other sequence classifiers by being one of the most efficient tools in this space. The algorithm, being essentially k-mer based, is very cheap to run, unlike full sequence alignment methods, such as BLAST.
However, Kraken 2 has been held back by the requirement that all data should fit in memory: you can not easily scale beyond the available RAM. In practice, it is hard to get machines with more than 1 or 2 TB of RAM, and so there has been a hard limit. With Slacken, we have demonstrated the ability to run arbitrarily large databases on commodity machines with 32-64 GB of RAM, thanks to our Apache Spark-based distributed architecture. There’s in principle no limit to how far this can go, and we are now planning for databases of 10-100 TB or more. The end goal will be to have the reliability and coverage of tools like BLAST, while keeping the low cost of Slacken.
However, in order to build such large databases while retaining specificity - a high fraction of species- and strain level classifications - something like our two-step classification method is necessary, in order to compensate for minimizers moving higher up in the taxonomy.
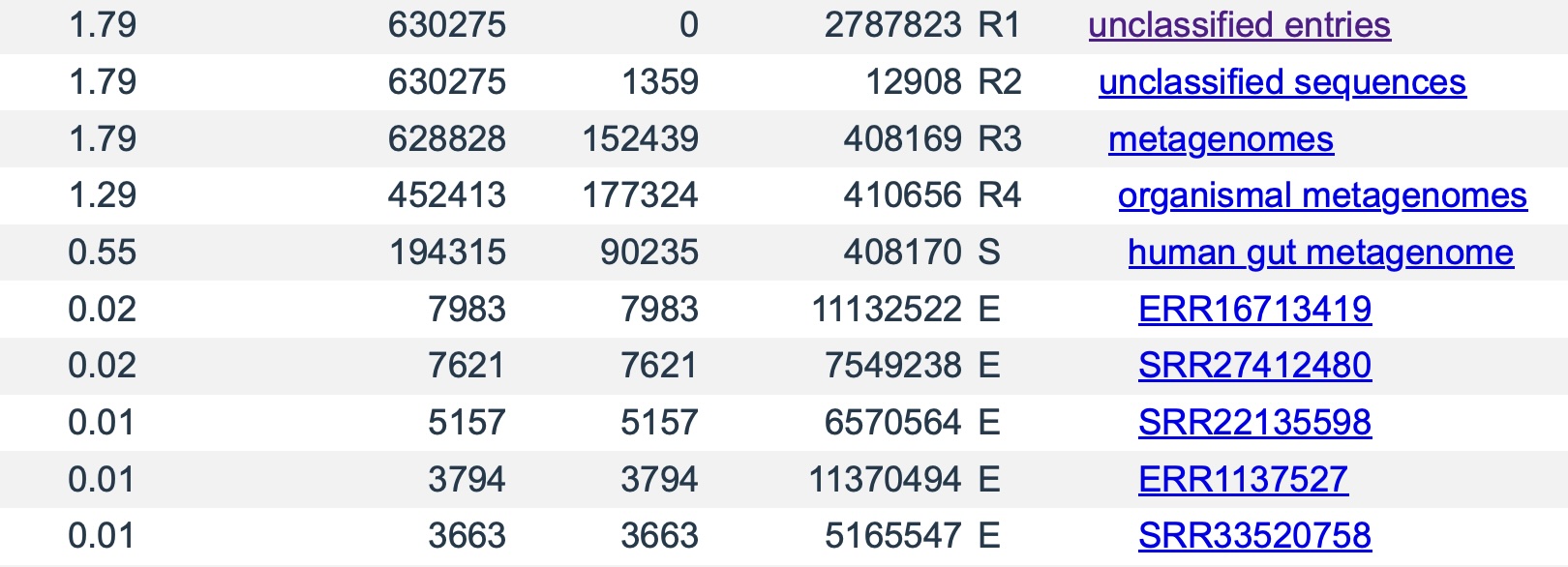
To build even larger databases, we are now going beyond reference genomes such as RefSeq (which Kraken 2 databases are typically based on) and exploring reference libraries built partly from WGS and even metagenomic WGS samples. In recent experiments, adding 5000 randomly selected SRA samples (for a total of 40 TB of NGS input data) to the RefSeq reference library yielded a 3 TB database. This increased classification results for a randomly selected test sample from 74% ( with the standard library) to 97%. The full results are available here (standard library) and here (RefSeq with 5000 SRA samples). The sample being classified was SRR31707655. In this context, reads from this sample will match other SRA samples if they share some novel DNA that was not part of a reference genome. The ability to match novel DNA in this way is what raises the classification rate.
This is currently a proof of concept, and we are now considering how to best design our reference libraries to get the most meaningful impact from this new technology.
We are actively looking for commercial or academic partners and collaborators to help explore the next stage of Slacken work and push the possibilities of metagenomic profiling. If you think this might be you, please get in touch with us at: info@jnpsolutions.io